
What is ISO 14971:2019 Risk Management for Medical Devices?
ISO 14971 addresses risk management and is the international standard designed for the medical device industry. This standard defines the best practices throughout the entire life cycle from design to distribution and maintenance. Additionally, ISO 14971 provides a thorough explanation of terms and definitions. It is paramount for your organization to guarantee that your products are safe and effective, and having a risk management system in place is crucial.
Today there are three versions of ISO 14971: ISO 14971:2007, EN ISO 14971:2012 and ISO 14971:2019. EN is the ISO standard for the European market. Everywhere else in the world ISO 14971:2019 remains the current standard. The EN version of the standard introduces three new annexes which have their own set of requirements. If you are doing business in the EU, then you should purchase this version of the standard. If you align your risk management process with the EU version, you will also meet the requirements for ISO 14971:2007.
|
|
|


Regardless of which standard you are looking at, the abstract describes both standards the same: “ISO 14971 is a key standard specifying a process for a manufacturer to identify the hazards associated with medical devices, including in vitro diagnostic (IVD) medical devices, to estimate and evaluate the associated risks, to control these risks, and to monitor the effectiveness of the controls. The requirements of this standard are applicable to all stages of the life cycle of a medical device.”
|
|
 Risk Management Procedure |

How are ISO 14971 and ISO 13485 related?
ISO 14791 and ISO 13485:2016 are related because they work together to create a QMS that is functional and addresses risk. ISO 13485 is focused on regulatory and customer requirements and for medical devices. As ISO 13485 requires risk analysis and record keeping pertaining to any risk, ISO 13485 looks to ISO 14971 for guidance. ISO 14971 is more detailed when it comes to risk management requirements.
The new revision of ISO 13485 expands risk management to include processes such as purchasing and training. Section 4.2.1 states that, “The organization shall apply a risk-based approach to the control of the appropriate processes needed for the quality management system.” In other words, anything that affects the quality system needs to be viewed from that risk perspective. Although this may not be new, it is a reminder that risk is a major part of your QMS and needs to be addressed to achieve ISO 13485:2016 certification.
Another tool to help manage risk is Failure Mode Effect Analyses (FMEA). This is a standard technique used to assess and evaluate potential risks in the design development phase, which continues during production process controls.
Top Management and Risk Management
The foundation of the risk management process must be top management. Top management has the responsibility for determining whether the product risks are acceptable or not. Further, they must be responsible for making sure there are adequate and appropriate resources for conducting risk management assessments and that these assessments are effective. They must ensure that the management processes are described, documented, and controlled as part of quality system procedures.
Risk Management is also multifaceted, and the responsibility of risk management should go beyond top management. Most organizations think of product developers and engineers taking on the majority of risk management roles beyond top management. Although developers and engineers play a pivotal role, risk management is a comprehensive process that requires all functional areas of the medical device company. This should encompass business development, marketing, manufacturing, sales, and end-users.
Common Risk Assessment Tools
There are many common tools for risk management, yet the regulatory guidance for exactly how to manage risk is intentionally vague allowing organizations to use whatever tools and processes necessary to manage risk.
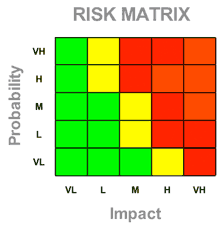
- Risk Matrix
- PHA = Preliminary Hazard Analysis
- FTA = Fault Tree Analysis
- FMEA = Failure Mode Effects Analysis
- HAZOP = Hazard Operability Analysis
- HACCP = Hazard Analysis and Critical Control Point
NOTE: Risk Management Worksheet Required for all operations or training and must be completed during the planning phase Reviewed before operations/training
Risk management is becoming front and central to every medical device regulatory agency. Today, regulatory agencies are even using risk-based processes throughout their internal processes when reviewing
ISO 14971 outlines a process to identify the hazards associated with medical devices. It helps ensure the safety of a medical device during the product’s life cycle. The process steps are:
- Estimate and evaluate risks
- Control risks
- Monitor risk control effectiveness
There are many risks associated with the design & production of Medical Devices and also in their use. Risk to safety of patients, users, handlers, and Regulatory Product liability must all be managed. Manufacturers must conduct and document a risk management process. As incorporating risk management into your organization can be difficult, here are two examples:
- Starting by benchmarking how other suppliers manage risks is greatly beneficial. Starting from a blank page can be daunting and is unnecessary. It is a great tactic to assess other similar suppliers’ risk management plan.
- Another key to successful risk management is to incorporate it throughout product realization. A strong risk management program starts with the initial customer contact and continues through the entire process. In the feasibility stage, the supplier considers the risk with such questions as: Do we have the right capabilities? Do we have the right machines? Can we order the right material? Do we have the ability to detect defects? Next, resources are evaluated. Do we have the resources to produce this part? Can we produce it at the volume needed? Risk is further managed during receiving by inspecting incoming materials. Another way to reduce risk is by evaluating suppliers prior to selection.
- FMEA is a common way to address risk, and we offer these resources:
Why Perform a risk analysis?
The top three reasons to perform a risk analysis are:
- It is required
- It could save costs of consequences
- Protection from product liability.
- Regulatory submissions checklists (PMA and 510k) used by the FDA include risk analysis.
Risk Management Plan in Your Organization
ISO 14971 provides a framework for a risk management plan for your organization as applied to medical devices. From initial analysis to risk control & evaluation, the probability and frequency of harm can be assessed, analyzed & managed.
Risk Analysis and Management Plan
When you create a management plan you need to address risk management and analysis. A mitigation plan should outline the risk management plan, the required resources, and the training/experience of those calculating the risks. Measure and monitor processes as required in ISO 13485.
A Risk Management Plan must include the following criteria:
- The organization must define the risk management activities and which products are included within that risk management plan. Multiple products may be able to be described within a single risk management plan.
- The organization must describe the intended use of the products.
- It is paramount to describe all risk management plans throughout the product life cycle.
- The risk management plan must describe the roles and responsibilities of personnel. Identification of the risk management team that will be reviewing and approving documents is also necessary to address.
- Define criteria for the product’s risk acceptability
- Define the methods to verify that the Risk Control measures are implemented and work.
- Describe how post-production information will be captured and fed into Risk Management activities.
Organizational Commitment
As required in ISO 13485, organizational commitment and involvement are essential. Risk objectives are part of the organization’s goals and resources must be allocated to properly mitigate risk.
Management commitment to risk analysis is necessary for medical device facilities. It is commonplace for Medical Device companies to believe that risk management should be in the hands of developers and engineers designing new products. Although these employees play an important role, medical device risk management is comprehensive and needs to be addressed throughout the organization in areas such as business development, marketing, manufacturing, sales, and end-users should be an integral part of your Risk Management process.
Estimate Risks
ISO 14971 requires medical device manufacturers to estimate risk by considering all possible negative consequences resulting from the use of their device. There are several ways to identify the risks in medical devices, then you estimate its probability and impact as defined in the standard.
Control Risk
Determine ways to minimize risk to an acceptable level, which must be clearly communicated to all stakeholders.
The Process
- Risk Management Planning
- Risk Analysis
- Risk Evaluation
- Risk Controls
- Overall Residual Risk Acceptability
- Risk Management Report
- Production & Post-Production Information
Three Rules of Risk Management:
The three main rules for risk management for ISO 13485:2016 are:
- Benefits must exceed cost
- Accept no unnecessary risk
- Make decisions at the appropriate level in the organization
ISO 14971 is Recognized Internationally
ISO 14971 is an international standard that is recognized universally. To give you an example of the importance of this standard the following governments recognize this standard:
- The U.S. Food and Drug Administration (FDA)
- The EU has harmonized with the European Medical Devices Directive 93/42/EEC.
- Australia TGA
- Japan MHLW
- ISO 13485 refers to ISO 14971 for guidance related to risk management.
MAKE ISO 13485 CERTIFICATION SIMPLE AND FOOLPROOF!

Our All-in-One Certification Package is a proven, efficient system. It gives you all you need to prepare for registration – in one simple to use package.