
What is ISO 13485?
The current version of the ISO 13485 standard is 13485:2016.
ISO 13485:2016 is the standard for a Quality Management System (“QMS”) for the design and manufacture of Medical Devices.
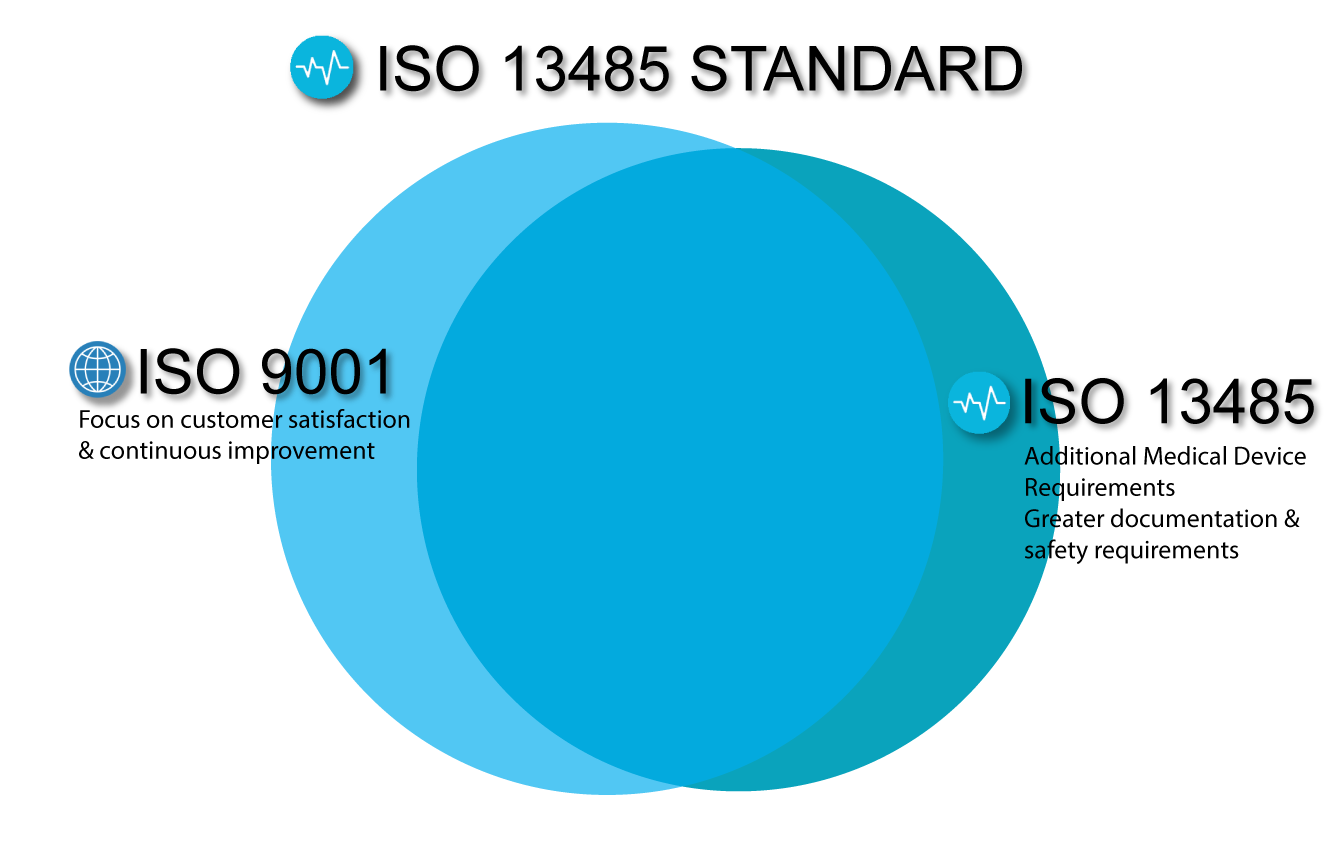
Certification to the standard requires an organization’s quality management system to pass a third-party Medical Device Single Audit Program, or “MDSAP” Audit. For the most part: ISO 13485 = ISO 9001 + Additional Medical Device Requirements.
While ISO 13485:2016 remains a stand-alone document, it aligns with ISO 9001:2008. (Yes, 2008, not 2015.) This is because it does not follow the high-level structure (Annex L) of the latest version of ISO 9001 (which is 9001:2015). In addition, the documentation and safety requirements are much greater under ISO 13485:2016, whereas ISO 9001:2015 puts focus on customer satisfaction and continuous improvement.
This global standard is mandatory in some countries, and in the U.S. the FDA has proposed a rule which would harmonize U.S. FDA 21 CFR 820 with ISO 13485:2016, making ISO 13485 the FDA’s mandatory QMS for Medical Devices (the rule is expected to be released in 2019). In the meantime, the medical device industry can rely on AAMI TIR102:2019, which is a bi-directional mapping tool that was released on August 30, 2019.
Purchase: AAMI TIR102:2019
U.S. FDA 21 CFR mapping to the applicable regulatory requirement references in ISO 13485:2016 Quality Management Systems.
A few details about ISO 13485:
ISO 13485 is the main Quality Management System (QMS) standard for medical devices, although several countries have their own set of regulations. As an example, the United States plans to harmonize the Food and Drug Administration (FDA) requirements for medical devices with ISO 13485. The scope of the QMS can be tailored for an organization, particularly in Section 7. The current version is ISO 13485:2016, which was published in March of 2016 (thus the: 2016).
- It does NOT matter what size your organization is: 1 person or 1 million people.
- It is NOT a standard for products. It does not define product quality. This is a process-based standard: you use it to control your processes, then your end product should meet the desired results.
- It is NOT a personal standard – a person cannot get certified to ISO 13485. Instead, an organization or company becomes certified. An individual, however, CAN become an ISO 13485 Certified Lead Auditor after a 5-day training course. This then allows them to audit other companies.
- It is NOT a membership group. An organization cannot “join” ISO 13485. To become ISO 13485 certified, your organization must:
- Follow the steps to implement an ISO 13485 quality management system.
- Then a Certification Body (CB or Registrar) audits the performance of your organization against the latest version of the ISO 13485 Requirements. If you pass this audit, the Registrar issues an ISO 13485 Certificate demonstrating that your organization is Registered to ISO 13485 for a three-year period. (See Who is able to grant certification.)
- Finally, the organization must be re-certified every three years in order to maintain its ISO 13485 certification status.
The Definition of ISO 13485 Certification
“ISO 13485 Certified” means an organization has implemented an ISO 13485 Quality Management System and has successfully met all of the requirements in ISO 13485. ISO 13485 evaluates whether your Quality Management System is appropriate and effective while emphasizing the safety and efficacy of medical devices.
MAKE ISO 13485 CERTIFICATION SIMPLE AND FOOLPROOF!

Our All-in-One Certification Package is a proven, efficient system. It gives you all you need to prepare for registration – in one simple to use package.
Some Helpful Resources:
- Learn the Steps to ISO 13485:2016 Certification
- Learn about the ISO 13485:2016 Requirements
- Learn about Global Medical Device Standards
- Learn about Medical Device Single Audit Program-MDSAP
- ISO 13485:2016 Training reviews all the clauses of ISO
- Compare ISO 9001:2015 and ISO 13485:2016
- Learn about the Benefits of ISO 13485:2016
History of ISO 13485 Revisions
Changes from ISO 13485:2003 to ISO 13485:2016
ISO 13485 was finally revised after 13 years and has many significant changes. The three main reasons for the updates are:
- The medical device regulatory environment has evolved since 2003.
- Risk management and risk-based decision-making processes have become the main focus of the entire medical device industry.
- ISO 13485:2016 no longer aligns with the current version of ISO 9001, but rather aligns with the previous revision, ISO 9001:2008.
For a more detailed discussion about these changes, go to our Compare ISO 13485:2016 to ISO 13485:2003 page.
What is ISO 1SO 13485:2012?
EN ISO 1SO 13485:2012 was a revision based off of ISO 13485:2003 established by the European Union. The EU has two standard organizations, the European Committee for Standardization (CEN) and the European Committee for Electrotechnical Standardization (CENELEC). When the CEN issues a standard based off of an ISO standard, you will see a prefix “EN”, and the year will be adjusted accordingly.
The standard includes parts of ISO 13485 and was a revision of ISO 13485:2003. It was created to be compliant with CE-marking requirements. The standard’s requirements remain unchanged, and the updated revision was created only for manufacturers placing devices on the market throughout Europe. ISO 13485:2003 remained the applicable standard for all other international markets, including the U.S.
What is ISO 13485:2003/Cor 1:2009?
ISO 13485:2003/Cor 1:2009 = ISO 9001:2008 + Medical Device Requirements
Technical Corrigendum 1 to ISO 13485:2003 was prepared by the ISO/TC 210 technical committee and published on January 8, 2009. It is a very brief document where the standard is aligned with ISO 9001:2000 and ISO 9001 is replaced with ISO 9001:2000 in pages v and vi at sub-clauses 0.3 and 0.4; in page 1 at sub-clause 1.1: and in page 25 at first paragraph of Annex B.
This document was published after the 2007 release of ISO 9001, making only minor adjustments. At the time, there were not enough adjustments to justify a new revision of the standard, but in 2016 ISO released a new revised standard ISO 13485:2016.
What is ISO 13485:2003?
ISO 13485:2003 = ISO 9001:2000 + Medical Device Requirements
ISO 13485:2003 Medical Devices – Quality Management System requirements for regulatory purposes is an ISO standard, originally published in 1996. These standard incorporated aspects of ISO 9001:2000 Quality Management System but is specific to the global medical device industry.