
ISO 13485 Documentation Requirements
How Much Documentation Do You Need?
Much depends on the size and complexity of your organization. You should have enough procedures to cover each section of the standard that applies to your business. Documented information is broken up into two types, documents, and records. A form is a kind of document. When the form is filled out it becomes a record. Quality manual, policy, procedure, or work instructions are other kinds of documents.
The specific documents and records you are required to control are listed in the table below.
The following ISO 13485 clauses require documented procedures to define and control the requirements and practices:
| Clause | Requirement |
| 4.1.1 | Roles undertaken by the organization under applicable regulatory requirements |
| 4.1.6 | Procedure and records for the validation of the application of computer software |
| 4.2.2 | Quality Manual |
| 4.2.3 | Medical device file |
| 4.2.4 | Procedure for document control |
| 4.2.5 | Procedure for record control |
| 5.3 | Quality policy |
| 5.4.1 | Quality objectives |
| 5.5.1 | Responsibilities and authorities |
| 5.6.1 | Procedure and records for management review |
| 6.2 | Procedure for training |
| 6.3 | Requirements for infrastructure and maintenance activities |
| 6.4.1 | Requirements for work environment |
| 6.4.2 | Arrangements for control of contaminated or potentially contaminated product |
| 7.1 | Process for risk management in product realization |
| 7.1 | Outputs of product realization planning |
| 7.2.2 | Records of the results of the customer requirements review and actions arising from it |
| 7.2.3 | Arrangements for communication with customers |
| 7.3.1 | Procedure for design and development |
| 7.3.2 | Design and development planning |
| 7.3.4 | Design and development outputs |
| 7.3.5 | Records of design and development review |
| 7.3.6 | Design validation plans, results, and conclusions |
| 7.3.8 | Procedure for transfer of design and development outputs to manufacturing |
| 7.3.9 | Procedure and records for control of design and development changes |
| 7.3.10 | Design and development file |
| 7.4.1 | Procedure for purchasing |
| 7.4.1 | Criteria and records for evaluation and selection of suppliers |
| 7.4.3 | Record of verification of purchased product |
| 7.5.1 | Record for each medical device or batch that provides traceability |
| 7.5.2 | Requirements for cleanliness of product |
| 7.5.3 | Requirements for medical device installation and acceptance criteria for verification of installation |
| 7.5.3 | Records for medical device installation and verification of installation |
| 7.5.4 | Procedure and records for servicing of the medical device |
| 7.5.5 | Records of sterilization process |
| 7.5.6 | Procedure and records of production and service provision process validation |
| 7.5.7 | Procedure and records for validation of process for sterilization and sterile barriers systems |
| 7.5.8 | Procedure for product identification |
| 7.5.9.1 | Procedure for traceability |
| 7.5.9.2 | Records of traceability and name and address of the shipping package consignee |
| 7.5.10 | Report on changes on customer property |
| 7.5.11 | Procedure for preserving the conformity of product |
| 7.6 | Procedure for monitoring and measuring |
| 7.6 | Record of calibration |
| 7.6 | Procedure and records for validation of the application of computer software used for monitoring and measuring |
| 8.2.1 | Procedure for customer feedback |
| 8.2.2 | Procedure and records for complaint handling |
| 8.2.3 | Records of reporting to regulatory authorities |
| 8.2.4 | Procedure for internal audit |
| 8.2.4 | Records of audits and their results |
| 8.2.6 | Identity of the person authorizing release of product |
| 8.3.1 | Procedure and record of control of nonconforming product |
| 8.3.4 | Records of rework |
| 8.4 | Procedure and records for data analysis |
| 8.5.2 | Procedure and records for corrective action |
| 8.5.3 | Procedure and records for preventive action |
Aside from the documents you will need for your individual jurisdiction, ISO requires you to document elements like:
- Quality policy and objectives
- Quality manual
- Computer software validation procedure
- Quality procedures and records
- Medical device file
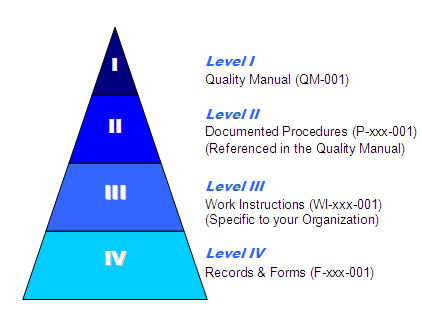
How do I set up my Documentation?
If you look at this pyramid, the lower the items are – the more specific they are to your business. The higher levels (QM, Proc) are dictated by the standard.
- The Quality Manual describes the quality management system
- Procedure describe a process (Purchasing)
- Work instructions describe an activity within a process (Creating a Purchase Order)
- Forms record the actions of an activity or process
- NOTE: A record is a completed form/Table/etc. that prove the action took place.
Do I need Procedures and Flow Diagrams?
No. Written procedures are more informative and much easier to edit than flows. Sometimes editing flowcharts can be tedious – you’re often better off creating them yourself.
You are required to have a diagram of your overall processes, and we recommend using software like Lucidchart to create them.
MAKE ISO 13485 CERTIFICATION SIMPLE AND FOOLPROOF!

Our All-in-One Certification Package is a proven, efficient system. It gives you all you need to prepare for registration – in one simple to use package.